Research
Our goal
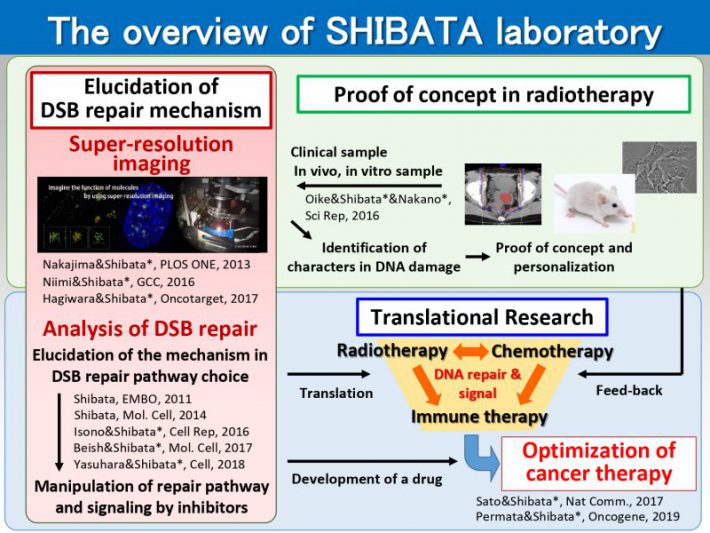
Our major goal is to elucidate the mechanism underlying the choice of DNA double-strand break (DSB) repair pathway. DSB is repaired via either non-homologous end joining (NHEJ) or homologous recombination (HR). Loss of the balance between NHEJ and HR causes genome instability. Because ionizing radiation (IR) is a potent DSB inducer, we are mainly investigating the mechanism of the choice of DSB repair pathway after X-ray (or carbon ion radiation in context of radiotherapy). In addition to radiotherapy, most chemotherapeutic agents kill cancer cells by causing DSBs. With the understanding of the molecular mechanism underlying the choice of repair pathway, we aim to manipulate DNA damage response and direct cancer cells toward cell death to improve the efficacy of cancer therapy.
Why do we need DNA repair research?
Following exposure to genotoxic stress, cells try to repair the DNA damage without introducing errors in DNA. For precise repair, cells correctly choose either NHEJ or HR for each situation. However, if cells select an inappropriate repair pathway due to the lack of balance between NHEJ and HR, it may cause genomic errors, i.e., mutations including deletion or chromosomal translocation.
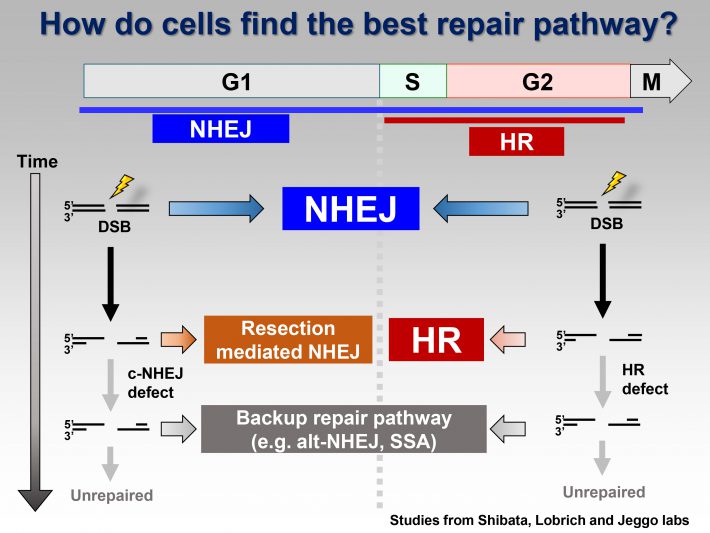
Repair process is always associated with DNA damage signaling. Cells are arrested by cell cycle checkpoint signals until the damage is repaired. For activating the cell cycle checkpoint machinery, the DSB ends activate ATM and downstream signal transducers. ATM is mainly activated at non-resected DSB ends, and ATR is activated at single-strand DNA (ssDNA) following DNA-end resection. Because resection requires 1–2 h after IR and ATM is not effectively activated at ssDNA, the signal transducer is switched from ATM to ATR at 1–2 h after DSBs.
The balance between ATM and ATR has to be well-orchestrated to organize the integrity in response to DNA damage; however, unbalanced DSB repair pathway, for example, no activation of ATR because of the loss of resection, results in the disorganization of cellular response after DNA damage.
1)Elucidation of the mechanism underlying DNA double-strand break repair pathway choice
Summary
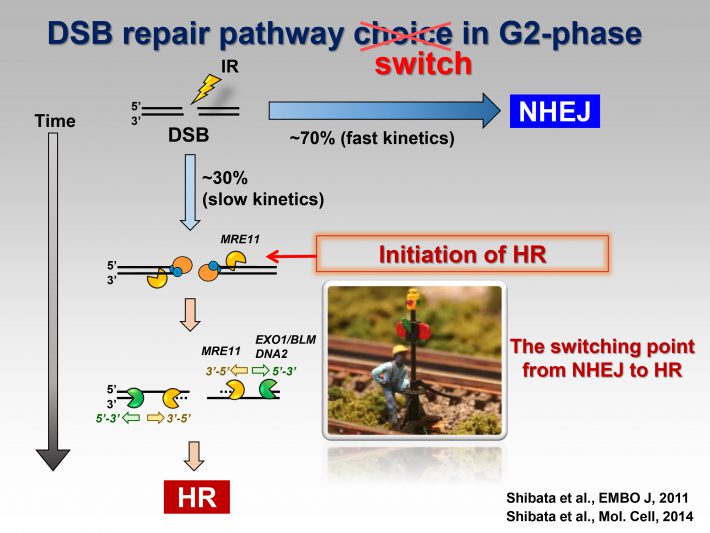
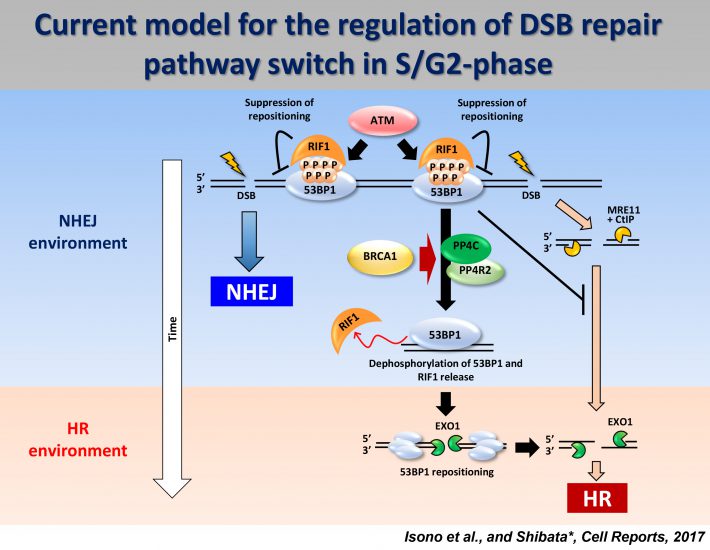
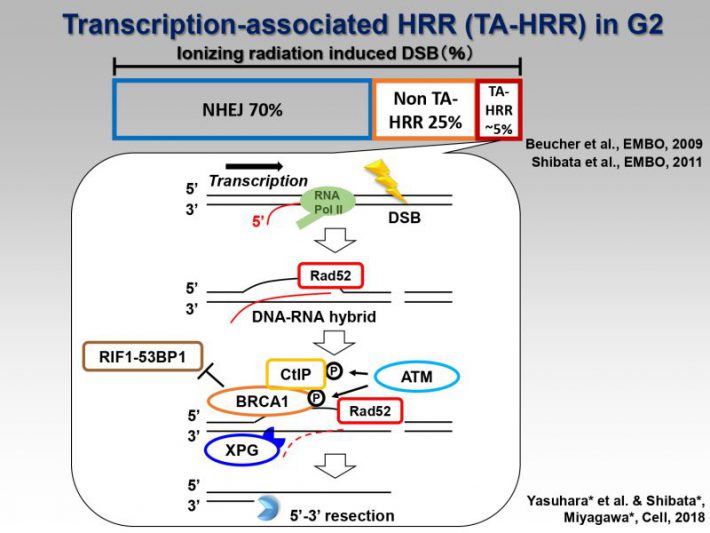
We propose a model for the switch of repair pathway from NHEJ to HR in S/G2 cells. Following the induction of DSBs, ATM phosphorylates 53BP1, even in S/G2 phase, at all the DSB sites followed by the transient recruitment of RIF1, which generates a pro-NHEJ environment. In G2, NHEJ factors rapidly repair ~70% of the DSBs. The remaining DSBs are not excessively resected due to the 53BP1 barrier in the presence of RIF1 from ~0.5-1 h post IR. MRE11 endonuclease initiates resection. When timely repair by NHEJ does not ensue, BRCA1 promotes 53BP1 dephosphorylation. PP4C plays a major role in 53BP1 dephosphorylation, which promotes RIF1 release. As resection proceeds, ATM activity is diminished. Thus, the failure to rephosphorylate 53BP1 combined with its dephosphorylation results in the release of RIF1. Removal of RIF1 allows 53BP1 repositioning. After removing the 53BP1 barrier, EXO1 can progress the elongation of resection required for HR.
Reference
- Factors determining DNA double-strand break repair pathway choice in G2 phase
EMBO J, 2011
http://www.ncbi.nlm.nih.gov/pubmed/21317870 - DNA Double Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities
Molecular Cell. 2014
http://www.ncbi.nlm.nih.gov/pubmed/24316220 - DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination.
Molecular Cell, 2017
https://www.ncbi.nlm.nih.gov/pubmed/28132842 - BRCA1 Directs the Repair Pathway to Homologous Recombination by Promoting 53BP1 Dephosphorylation
Cell Reports, 2017
https://www.ncbi.nlm.nih.gov/pubmed/28076794 - [NEW!!] Human Rad52 Promotes XPG-Mediated R-loop Processing to Initiate Transcription-Associated Homologous Recombination Repair.
Cell, 2018
https://www.ncbi.nlm.nih.gov/pubmed/30245011
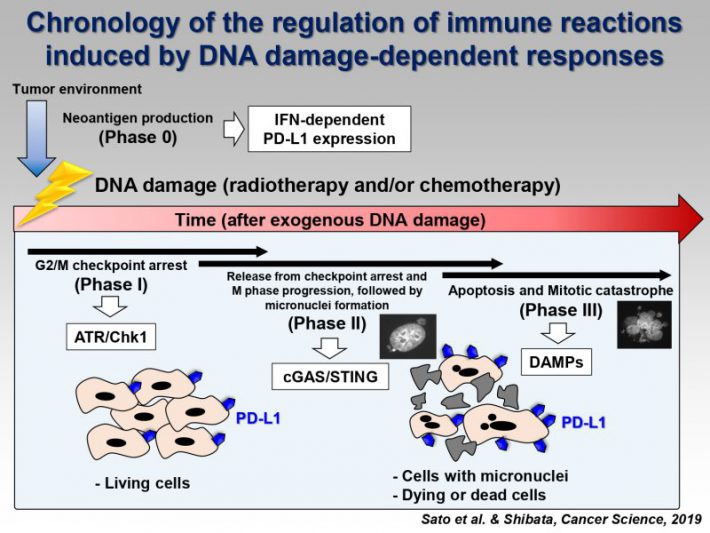
2)Investigating the molecular mechanism underlying PD-L1 expression after DNA damage for precision radio-immunotherapy
Summary
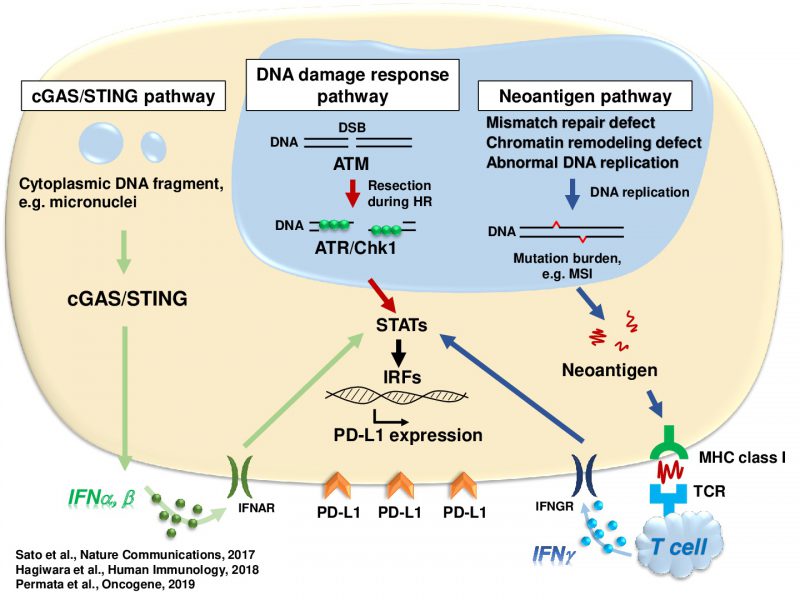
Accumulating evidence suggests that exogenous cellular stress induces PD-L1 upregulation in cancer. A DNA double-strand break (DSB) is the most critical type of genotoxic stress, but the involvement of DSB repair in PD-L1 expression has not been investigated. Here we show that PD-L1 expression in cancer cells is upregulated in response to DSBs. This upregulation requires ATM/ATR/Chk1 kinases. Using an siRNA library targeting DSB repair genes, we discover that BRCA2 depletion enhances Chk1-dependent PD-L1 upregulation after X-rays or PARP inhibition. In addition, we show that Ku70/80 depletion substantially enhances PD-L1 upregulation after X-rays. The upregulation by Ku80 depletion requires Chk1 activation following DNA end-resection by Exonuclease 1. DSBs activate STAT1 and STAT3 signalling, and IRF1 is required for DSB-dependent PD-L1 upregulation. Thus, our findings reveal the involvement of DSB repair in PD-L1 expression and provide mechanistic insight into how PD-L1 expression is regulated after DSBs.
Reference
- Inhibition of the HDAC/Suv39/G9a pathway restores the expression of DNA damage-dependent major histocompatibility complex class I-related chain A and B in cancer cells.
Oncology Reports, 2017
https://www.ncbi.nlm.nih.gov/pubmed/28677817 - DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells.
Nature Communications, 2017
https://www.ncbi.nlm.nih.gov/pubmed/29170499 - Analysis of programmed death-ligand 1 expression in primary normal human dermal fibroblasts after DNA damage.
Human Immunology, 2018
https://www.ncbi.nlm.nih.gov/pubmed/29859207
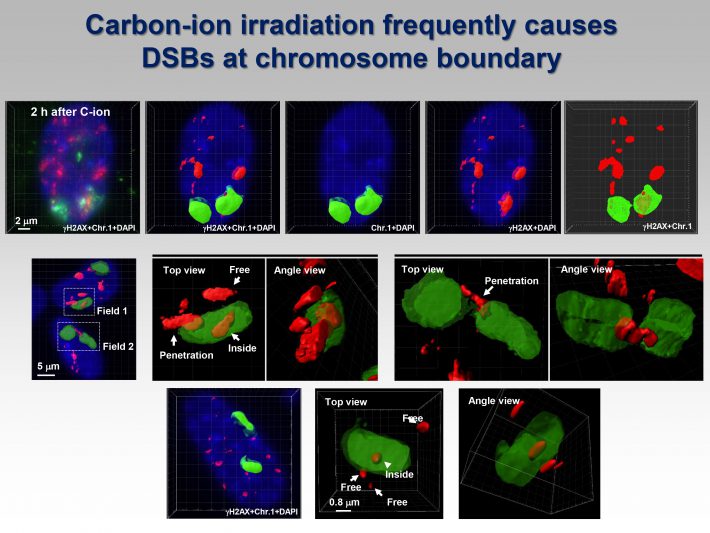
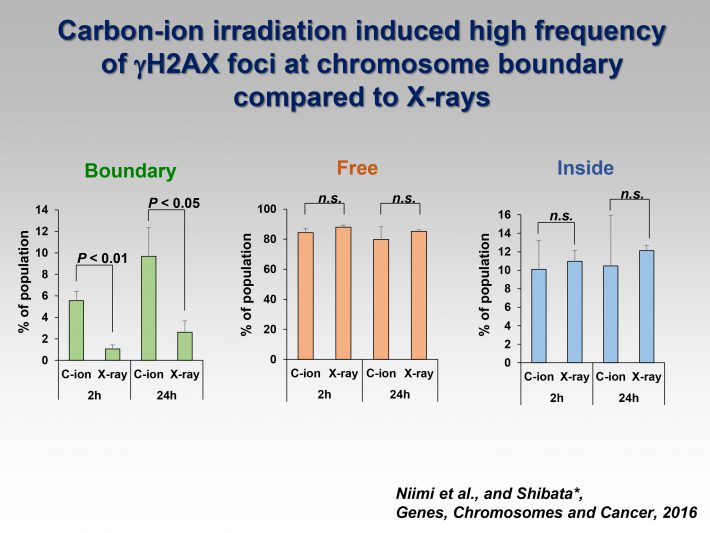
3)Visualization of clustered DNA double strand break repair after high LET carbon ion irradiation.
Summary
Photon and particle irradiation causes different types of DNA damage. Photons, such as X- or -rays, induce DNA damage with a random distribution because of low-density energy deposition. In contrast, particle radiation with high linear energy transfer deposits high-density energy, resulting in the formation of clustered DNA damage containing DNA double- and single-strand breaks as well as base damage. To date, accumulated evidence has revealed that heavy-ion radiation results in dynamic chromosomal aberration, e.g., chromosomal translocations or large deletion mutations; however, the high incidence of these mutations after heavy-ion radiation cannot be simply explained by the formation of clustered DNA damage. Currently, we have investigated the unique characteristics of DNA damage induced by heavy-ion radiation based on our recent findings using high-resolution imaging.
Reference
- Visualisation of gH2AX Foci Caused by Heavy Ion Particle Traversal; Distinction between Core Track versus Non-Track Damage
PLOS ONE, 2013
http://www.ncbi.nlm.nih.gov/pubmed/2396707 - Visualization of complex DNA double-strand breaks in a tumor treated with carbon ion radiotherapy
Scientific Reports, 2016.
http://www.ncbi.nlm.nih.gov/pubmed/26925533 - Identification of DNA Double Strand Breaks at Chromosome Boundaries Along the Track of Particle Irradiation
Genes, Chromosomes and Cancer, 2016.
http://www.ncbi.nlm.nih.gov/pubmed/27113385 - 3D-structured illumination microscopy reveals clustered DNA double-strand break formation in widespread γH2AX foci after high LET heavy-ion particle radiation.
Oncotarget, 2017
https://www.ncbi.nlm.nih.gov/pubmed/29312614